
Transitioning from Startup to Close: The Lifecycle of Clinical Project Management
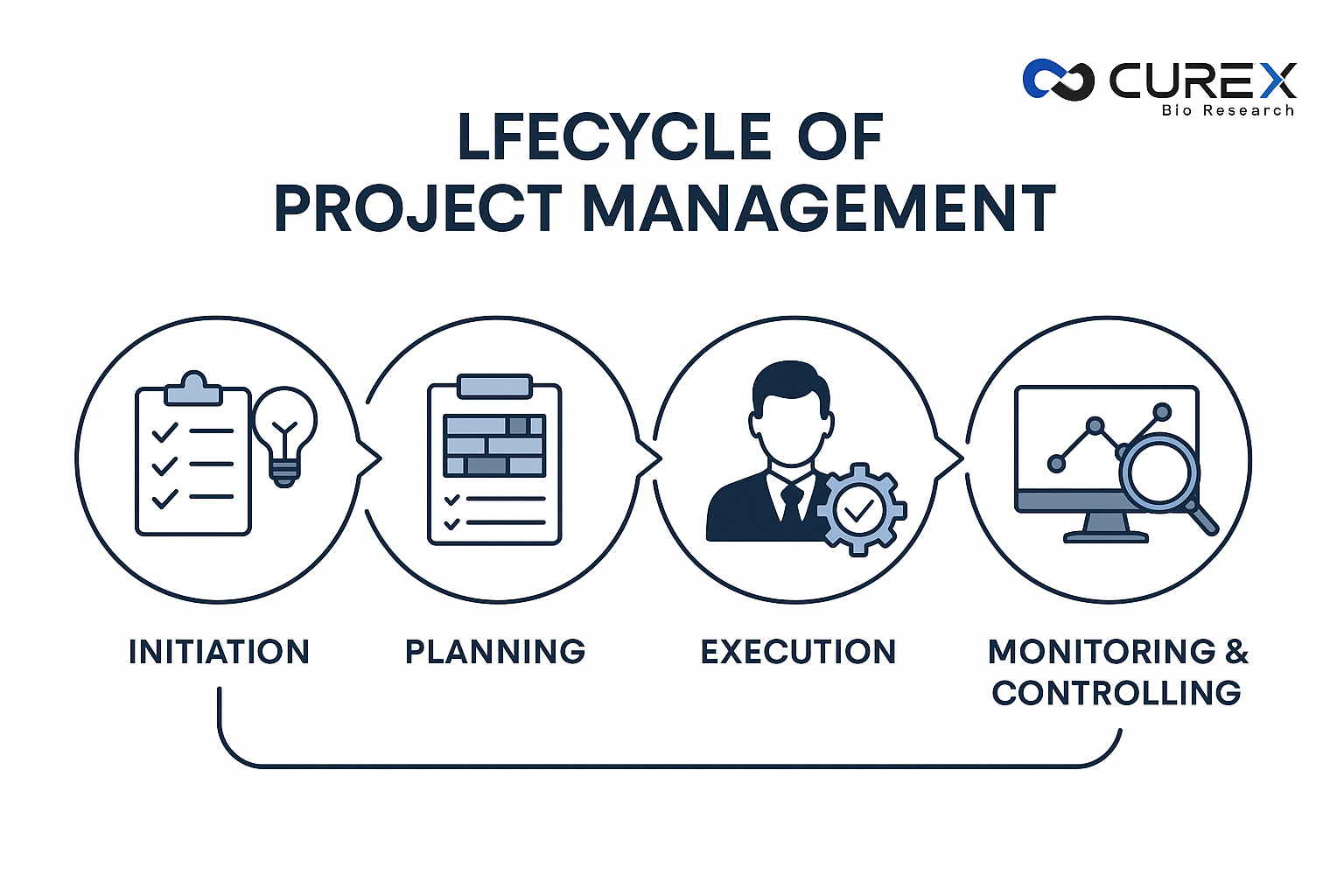
Clinical research relies heavily on effective clinical project management for the successful execution of trials. The lifecycle, outlined in the Project Management Body of Knowledge (PMBOK), comprises five phases: initiation, planning, execution, monitoring and controlling, and closing. CUREXBIO supports sponsors through this entire lifecycle using innovative tools and data-driven strategies that streamline operations and improve outcomes. Strong Clinical Project Management ensures that each phase of a clinical trial is strategically aligned with scientific goals, regulatory expectations, and patient safety standards—allowing sponsors to execute projects efficiently and with greater predictability. Initiation The initiation phase marks the start of a project by defining objectives, assessing feasibility, identifying stakeholders, and outlining scope. This stage includes developing a project charter, conducting a needs assessment, and forming the core team.Example: Imagine a biotech company developing a new oncology drug. During initiation, the project manager (PM) drafts a charter specifying the trial’s goal—to evaluate the drug’s efficacy in Phase II patients with specific cancer types. CUREXBIO helps stakeholders align timelines, budgets, and compliance requirements, performing gap analyses to ensure adherence to FDA and ICH guidelines from the very beginning. Strategic Outlook (Planning) The planning phase involves creating detailed roadmaps for scope, schedule, budget, risks, and quality. Tools like Gantt charts and risk registers help define deliverables and assign responsibilities.Example: The oncology trial’s plan includes site selection, patient recruitment, and data collection over 18 months. Risks like IRB approval delays are identified early, with contingency plans ready. Budgets are allocated for CRO services, investigational supplies, and monitoring visits. CUREXBIO integrates digital platforms that enhance planning efficiency and strengthen overall clinical project management. Execution Execution is the action-oriented phase where teams bring the plan to life through site activation, patient enrollment, data collection, and cross-functional coordination.Example: In the oncology study, the PM oversees training, drug shipment, and patient screening while addressing challenges like low enrollment through targeted recruitment strategies. At CUREXBIO, experienced project leads ensure smooth execution and proactive issue management, keeping trials within timelines and scope. Monitoring and Controlling This phase runs alongside execution, focusing on tracking progress, managing risks, and maintaining quality. Key performance indicators (KPIs) are used to evaluate performance and initiate corrective actions when needed.Example: CUREXBIO’s approach to clinical project management blends centralized and risk-based monitoring, helping detect issues early, maintain data integrity, and ensure compliance with study protocols. Wrapping Up and Learning Lessons (Closing) The closing phase ensures that all project activities are completed as planned, including data lock, final reporting, and documentation archiving. It also involves reviewing lessons learned for future improvements.Example: Once the trial concludes, the PM finalizes regulatory submissions, holds close-out meetings, and ensures proper knowledge transfer. CUREXBIO supports teams during closure by managing regulatory filings and documentation, ensuring a seamless transition to post-marketing studies. Overview of the Lifecycle Phase Key Activities Example Outcomes Challenges Initiation Define scope, charter, stakeholders Approved project charter Misaligned expectations Planning Develop schedules, budgets, risk plans Comprehensive management plan Overly optimistic timelines Execution Implement activities, manage teams Patient enrollment and data collection Resource shortages Monitoring & Controlling Track performance, manage changes Corrective actions and audits Deviation from plan Closing Finalize deliverables, lessons learned CSR submission and archiving Incomplete documentation This structured lifecycle ensures projects transition smoothly from startup to close, minimizing risks and maximizing efficiency through sound clinical project management practices. Managing the Lifecycle of Clinical Project Management with CUREXBIO CUREXBIO provides tailored solutions for every stage of clinical project management, from feasibility to regulatory submission and beyond. With experienced, multidisciplinary global teams and advanced, proven scientific methodologies, we help sponsors execute trials efficiently, compliantly, and on schedule—empowering innovation in clinical research.
Risk-Based versus Traditional Monitoring: How Site Monitoring Services Are Evolving

In the ever-evolving landscape of clinical trials, site monitoring services play a pivotal role in safeguarding data quality, patient safety, and regulatory adherence. For decades, the gold standard has been traditional on-site monitoring, where clinical research associates (CRAs) conduct frequent in-person visits to trial sites. However, with the rise of digital tools, big data analytics, and guidance from regulatory bodies like the FDA and EMA, the industry is shifting toward more efficient approaches. This blog explores the key differences between classical on-site monitoring and modern alternatives—risk-based monitoring (RBM), centralized monitoring, and hybrid models. At CUREXBio, we’re at the forefront of these changes, helping sponsors optimize their trials for better outcomes through advanced site monitoring services in India. Traditional On-Site Monitoring services: The Tried-and-True Approach Classical on-site monitoring involves regular visits by Clinical Trial Investigators (CRAs) to clinical trial sites to verify source data, review records, and ensure protocol compliance. This method, which includes 100% source data verification, allows direct interaction with site staff, immediate issue resolution, and a comprehensive understanding of site operations. Advantages: Establishes solid relationships between CRAs and site staff. Facilitates immediate identification and resolution of problems like protocol deviations or adverse events. Supplies thorough oversight, especially useful in complex or high-risk trials. Disadvantages: Resource requirements: Travel expenses, time, and staff needs can increase trial budgets by 20-30%. Variation from low-risk sites or data points prompts wasted work. Scalable across global, multi-site trials, especially in disruption like a pandemic. While efficient, this model has received critiques for being “one size fits all,” resulting in calls for more tailored approaches. Risk-Based Monitoring (RBM): Focusing on What Matters Most Risk-based monitoring prioritizes resources based on identified risks using data analytics. Sponsors assess site performance, data variability, and protocol complexity to determine monitoring intensity. High-risk sites receive frequent on-site visits, while low-risk ones rely on remote reviews. This aligns with ICH E6(R2) guidelines for quality management Advantages Cost efficient: Decreases site visits by 30-50% and reduces total costs. Increases data quality by analyzing inappropriate potential risks that are critical, which reduces Increases efficiency via the availability of dashboards to actively monitor risks and conduct data review. Disadvantages Necessitates solid data systems and appropriate training for proper risk appraisal. May overlook some emerging risks if the initial assessment is incorrect. Less interaction in person, potentially impacting relationships at the site. RBM is a data-centric evolution of trial management that makes it more responsive and adaptive. Centralized Monitoring: Remote Oversight from Afar Centralized monitoring is a method used by sponsor teams or representatives to conduct analytical reviews from a central location, utilizing statistical methods, key risk indicators, and electronic data capture systems to detect anomalies, trends, or inconsistencies without physical visits. Advantages: Scaling for large trials: Great at dealing with significant amounts of data as a result of automation and AI. Gap finding: Spotting systemic issues across sites (e.g. outliers in the data, fraud signal, etc.) Decreased air travel: Doing less air travel and/or reducing environmental impact Disadvantages: Limited to data on hand: Cannot address issues that require being on-site to verify or calibrate equipment, etc. Reliant on technology: Reliant on reliable data system technology and integration of technology. Lacking site specific detail that humans on site can observe. Centralized monitoring excels in complementing other methods, providing a bird’s-eye view of trial health. Hybrid Models: The Best of All Worlds Hybrid monitoring is a flexible strategy that combines on-site, risk-based, and centralized approaches to create a tailored approach. It includes centralized routine data reviews, targeted on-site visits, and remote monitoring, often incorporating remote source data verification and virtual tools. Advantages Cost-effective efficiency: Provides good value for money while providing oversight and might improve oversight by 20-40%. Versatile: Can accommodate all stages of a trial from early phase to post-marketing. Improved compliance: Blends real-time data with scheduled in-person interactions. Disadvantages Challenge of implementation: Requires alignment teams and operationalizing clear protocols. Costlier upfront facility for linked systems. Risk of inconsistency when transitioning from one approach to another. Hybrid models are being used in studies more frequently, especially with decentralized or virtual studies. Comparing the Models: A Side-by-Side Overview of site monitoring services Aspect Traditional On-Site Risk-Based (RBM Centralized Hybrid Focus Comprehensive SDV across all sites Targeted on high-risk areas Data analytics from central location Combination of all, based on needs Visit Frequency Regular, fixed schedule Variable, risk-dependent Minimal to none on-site Mix of on-site, remote, and central Cost Efficiency Low (high travel costs) High High | Medium-High Data Review Manual, in-person Data-driven prioritization Statistical and automated Integrated multi-method Scalability Limited for global trials Good Excellent Excellent Regulatory Alignment Standard but outdated Aligned with ICH E6(R2) Supports RBQM Flexible and compliant Patient Safety Impact Direct oversight Proactive risk mitigation | Early trend detection Comprehensive coverage This table underscores how modern models address the limitations of traditional methods while enhancing overall trial quality. The Evolution of Site Monitoring services : Toward a Smarter Future The shift from traditional to advanced site monitoring services reflects broader industry trends, including digital transformation and the lessons from events like COVID-19, which accelerated remote and hybrid adoption. By 2025, it’s estimated that over 70% of trials will incorporate RBM or centralized elements, driven by cost savings and improved outcomes. At Curex Bio, we specialize in site monitoring services, offering customized monitoring solutions that ensure compliance, efficiency, and innovation. Our expert teams use cutting-edge tools to deliver seamless hybrid models, helping you navigate complex trials with confidence. Whether you’re planning a new study or optimizing an ongoing one, embracing these modern approaches can transform your clinical operations. Contact CUREXBIO today to learn how we can support your site monitoring needs.
Navigating the Future of Drug Safety: Pharmacovigilance Solutions with CUREXBIO
Pharmacovigilance (PV) is crucial in the fast-paced pharmaceutical development world, as it ensures drug safety and human lives. CUREXBIO is at the forefront of transforming challenges into opportunities for innovation and compliance, ensuring the safety of new therapies and reducing the risk of adverse events, drug interactions, and side effects. This blog discusses the complexities of pharmacovigilance and drug safety solutions, focusing on CUREXBIO’s tailored services that enable pharma companies worldwide to accelerate safer drug launches, addressing the challenges faced by those in clinical research, drug development, or regulatory affairs. What is Pharmacovigilance? The Backbone of Drug Safety PV is the science of detecting, assessing, understanding, and preventing adverse effects or drug-related problems, extending beyond clinical trials to the entire drug lifecycle, from preclinical stages to post-marketing surveillance. Why does it matter? Robust PV systems can reduce adverse drug reactions by up to 30% by implementing proactive risk management, including adverse event reporting and signal detection. These solutions ensure therapies are effective and safe for diverse patient populations, reducing the annual number of reported adverse reactions. PV is crucial in understanding drug behaviour, interactions with other medications, and risk mitigation before patient impact. Without it, promising drugs could face recalls, lawsuits, or even harm to users, highlighting the importance of PV. Shifting Dynamics of Pharmacovigilance The pharma landscape is increasingly complex due to globalization, regulatory changes from the FDA and EMA, and the rise of biologics, gene therapies, and personalized medicine. The challenge of identifying actionable safety signals in real-world data from wearables and electronic health records is immense. Drug safety solutions are integrated platforms and services that use AI, analytics, and expert oversight to streamline compliance, improve reporting accuracy, and minimize risks, enabling faster therapies to market without compromising safety. How CUREXBIO Revolutionizes Pharmacovigilance At CUREXBIO, a global leader in clinical research organization (CRO) services with hubs in India, the USA, and Canada, we’re passionate about bridging scientific insights with real-world impact. Our PV arm is integrated into their end-to-end drug development ecosystem, specializing in clinical trials and safety monitoring. With a team of PV experts, they help clients navigate the PV maze with confidence. Here’s how CUREXBIO helps elevate your drug safety game: Customized Preclinical and Pharmacokinetic Support We conducts specialized exploratory DMPK studies to understand a drug’s absorption, distribution, metabolism, and excretion profile, providing crucial insights for human trials. We also perform pharmacokinetic (PK) drug profile analysis, including dose selection, exposure assessment, and drug-drug interaction forecasting, using animal data. By linking of PK data to toxicology outcomes helps correlate exposure levels with potential toxic effects, justifying safe dosing and creating a comprehensive safety narrative from the start. End-to-End CRO Services for Seamless Integration At CUREXBIO, we provides clinical trial protocol development with PV best practices, ensuring safety throughout phases, and offers post-marketing surveillance using streamlined reporting for real-time signal detection. Tech-Driven Efficiency and Global Compliance Our unique approach is our commitment to advanced safety data systems and cutting-edge technologies, ensuring timely, accurate deliverables that meet global standards. Our in-house team of seasoned PV professionals ensures high-quality outputs without outsourcing pitfalls, ensuring high-quality outputs. CUREXBIO’s Outcome driven advantage Risk Minimization: Proactive PK-tox linking reduces surprises in later stages, potentially slashing development costs. Rollout Speed: Tech-enabled workflows cut reporting times, helping you hit regulatory deadlines. Patient-Centric Safety: Every service prioritizes subject well-being, fostering trust and ethical innovation. Adaptable Solutions: Whether you’re running a Phase I trial or global surveillance, our flexible models adapt to your needs. We’ve helped clients transform raw data into market-leading therapies, supporting everything from adverse event documentation to regulatory submissions. Contact us on bd@curexbio.com for more our PV services.
Mastering the Clinical Development: Creating and Optimizing Your Plan with CUREXBIO

A well-designed clinical development plan (CDP) is crucial in drug development, guiding pharmaceutical companies from preclinical testing to regulatory approval. CUREXBIO understands the intricacies of this process and is dedicated to guiding companies through every step. A robust CDP minimizes risks, optimizes resources, and accelerates time-to-market, making it essential for start-ups and established firms in the drug development process. This blog highlights the process of creating and optimizing a clinical development plan, highlighting CUREXBIO’s expertise in clinical trials, pharmacokinetics, and regulatory strategy, and how they help clients refine these plans for maximum impact. Understanding the Clinical Development Plan A clinical development plan is a comprehensive roadmap that integrates scientific, regulatory, and operational aspects to showcase a drug’s safety, efficacy, and quality. Ensuring Patient Safety: Structured trials enable early identification of potential issues, facilitating timely interventions through systematic risk assessment. Proving Efficacy: It ensures the design of trials accurately measures therapeutic benefits, addressing any data gaps. Navigating Regulations: A robust CDP, in line with global standards like FDA and EMA, facilitates approvals and minimizes delays. Resource Optimization: The efficient allocation of time, budget, and personnel is a key factor in enhancing the overall success of a program. Partnering with experts like CUREXBIO is crucial for development programs to avoid costly setbacks, regulatory hurdles, and failure, transforming potential pitfalls into strategic advantages. How to Create a Clinical Development Plan A CDP requires collaboration among clinical, regulatory, and statistical experts, and a step-by-step guide is provided for creating one. Target Identification: Start by pinpointing the disease or condition your drug aims to treat, considering unmet needs and market potential. Preclinical Testing: Conduct animal studies to evaluate safety, efficacy, and dosing. This data informs human trial designs and helps predict pharmacokinetics. Biomarker Identification (If Applicable): Select biomarkers for diagnostics, prognostics, pharmacodynamics, or safety monitoring to enhance trial precision. Phase I Trial Design: Define participant numbers, dose escalations, and endpoints. Collaborate with pharmacologists to model optimal dosing. Phase II Trial Design: Build on Phase I results to refine efficacy endpoints, and safety assessments, incorporating preclinical insights. Phase III Trial Design: Scale up with larger cohorts, focusing on endpoints that support labeling claims and demonstrate clear benefits. Data Analysis Plans: Establish statistical methods and quality controls to ensure robust, interpretable results. Regulatory Interactions: Outline filings, meetings, and strategies for engaging authorities like the FDA or EMA. Safety Monitoring: Develop protocols for ongoing vigilance during trials and post-approval. Optimization of Clinical Development Plan Optimization turns a good plan into a great one, reducing costs, enhancing flexibility, and improving outcomes. Follow these strategies: Complete Review: Analyze existing data, regulatory changes, and program goals to spot improvement areas. Expertise Consultation: Engage specialists in clinical, regulatory, and stats for tailored advice. Improve Target Population: Use demographics and disease data to focus on responsive subgroups. Maximize Trial Design: Adjust sample sizes, arms, and endpoints for better efficiency. Incorporate Real-World Evidence (RWE): Use external data to refine designs and expand insights. Enhance Regulatory Strategy: Collaborate with agencies on endpoints and benefit-risk assessments. CUREXBIO Elevates Your Clinical Development Journey Our team of experts in pharmacokinetics, toxicology, and regulatory affairs provides comprehensive support for clinical development plans, ensuring clients create and optimize successful plans. Preclinical and PK Expertise: We provide exploratory DMPK studies and pharmacokinetic profiling to establish a robust foundation, linking exposure data to safety outcomes for informed dosing. Trial Design and Execution: From Phase I to III, focus on protocol development, adaptive designs, and biomarker integration to ensure efficient and compliant trials. Regulatory and Safety Integration: We ensure safety through robust monitoring, and incorporating real-world relevance of RWE. Data-Driven Optimization: Utilizing advanced analytics and AI, we streamline plans based on interim data, reducing risks and expediting approvals. We’re not just service providers, we’re collaborators in your quest for ground-breaking therapies. Contact us for our robust and futuristic services on bd@curexbio.com
Understanding Data Security and Compliance of Clinical Data Management

In today’s fast-evolving landscape of clinical research, where digital platforms and remote operations are becoming the norm, prioritizing compliance and data protection is more crucial than ever. This blog discusses the basics for trial compliance, importance of GDPR, HIPAA and CDSCO role in data integrity along with focus on clinical data management. It emphasizes the need to maintain participant well-being, maintain findings reliability, and build stakeholder confidence. Comprehending the Essentials of Trial Compliance At its core, compliance in clinical studies involves following relevant laws, moral standards, and organizational rules from start to finish. This approach guarantees that investigations are carried out with integrity, respect for ethics, and results that hold up under examination. Major guidelines to consider encompass: Figure 1: Basics of Trial Adherence Beyond these, it’s essential to align with in-house procedures, obtain ethical board endorsements, and meticulously record all actions in the study. The Role of Data Protection in Research Data protection means using strategies and technologies to protect private information—information about study participants is at the core of this work—from being misused, altered, or lost. In the instance of a study, this means protecting forms for case studies, consent information, medical histories, test results, and access for program staff, etc. Strong data protection will meet legal obligations, but it can also prevent violations and maintain confidentiality. Fundamental Standards for Trial Adherence To align with oversight expectations, organizers and locations should establish key safeguards: Figure 2: Fundamentals and Key Elements for Trial Compliance Consistency with global practices: Concerning consent procedures, result reliability, oversight and incident experience. Compliantwith the electronic rules: Including safe approvals, system checks and actionstraces. Compliancewithprivacy laws: Protection for health and personal information, includingconsent for use and disclosure. Ethical review process: Review of proposals, consents, and ongoing reviews.. Keeping thorough records is key—authorities will demand proof of education, clearances, handling methods, and any variations from the plan. Vital Protection Strategies for Digital Studies Figure 3: Strategies for Clinical Data Management Encoding information: Secure transfers and storage with strong protocols. Permission-based entry: Restrict access according to duties and positions. Safe cloud setups: Opt for reliable providers with logging features and location-specific storage. Recovery plans: Routine saves and drills for restoring operations. Activity records: Track user actions automatically for future reviews. The GDPR (General Data Protection Regulation),, HIPAA (Health Insurance Portability and Accountability Act), and CDSCO (Central Drugs Standard Control Organization) guidelines are crucial for handling sensitive patient data in clinical research. Each focuses on data protection, privacy, and compliance, tailored to their respective regions and regulatory environments. These guidelines are essential for effective Clinical Data Management and safeguarding patient data and ensuring ethical, secure clinical trials, providing insights from their principles. Importance of GDPR in Handling Sensitive Patient Data The GDPR, enforced in the European Union since 2018, sets stringent standards for processing and protecting personal data, including sensitive health information used in clinical trials. Figure 4: GDPR Mandates Protecting Participant Rights GDPR safeguards participant rights, ensuring they can control their health data usage in clinical trials, thereby fostering trust and ensuring the protection of their privacy. Consent and Transparency: GDPR mandates clear, informed consent for data collection and processing, requiring participants to be informed about the purpose, scope, and duration of data use, in line with ethical standards. .Data Minimization and Security: GDPR mandates data collection and security measures like encryption and pseudonymization to minimize breaches, especially in sensitive health data like eCRFs or lab results. Cross-Border Data Transfers: GDPR governs data transfers outside the EU for multinational trials, requiring safeguards like Standard Contractual Clauses to maintain consistent protection levels. Penalties for Non-Compliance: GDPR violations can result in fines of €20 million or 4% of annual global turnover, making compliance crucial to prevent financial and reputational damage. Importance of HIPAA in Handling Sensitive Patient Data HIPAA, a U.S. regulation, focuses on protecting Protected Health Information (PHI) in healthcare and research settings. Figure 5: Importance of HIPAA Safeguarding PHI: HIPAA outlines standards for securing PHI, including medical histories and test results, in clinical trials, ensuring confidentiality through eCRFs, eConsent, and health records. Access Controls and Accountability: HIPAA mandates role-based access control (RBAC) and audit trails to monitor PHI access and prevent unauthorized access, a crucial risk highlighted in the document. Patient Privacy Rights: HIPAA allows patients to access their PHI and request restrictions on its use, ensuring ethical data handling in trials, similar to GDPR’s participant-centric approach. Breach Notification and Vendor oversight: HIPAA mandates prompt notification of data breaches to protect participants and comply with regulations. It also requires Business Associate Agreements (BAAs) with vendors to ensure they meet security standards, reducing the impact of incidents like those mentioned in the document. Importance of CDSCO Guidelines in Handling Sensitive Patient Data The CDSCO, India’s drug regulatory authority, oversees clinical trials under the Drugs and Cosmetics Act, 1940, and the New Drugs and Clinical Trials Rules, 2019. Its guidelines address data protection and ethical conduct in India’s growing clinical research sector. Figure 6: CDSCO Framework for Clinical Data Management Ethical Data Handling: CDSCO mandates adherence to ethical guidelines, including Schedule Y and ICH-GCP principles. Requiring informed consent and data confidentiality. Informed Consent Requirements : CDSCO requires detailed, culturally appropriate consent processes, ensuring participants understand how their data (e.g., health records, lab results) will be used and protected. Data Integrity and Security: CDSCO guidelines stress maintaining data integrity through validated systems and secure storage, aligning with the document’s call for encryption, audit trails, and GxP-compliant platforms. Local Oversight: CDSCO requires Ethics Committee approvals and regular monitoring to ensure data protection practices are followed, similar to the document’s IRB/ethics oversight requirements. Adverse Event Reporting: CDSCO mandates prompt reporting of adverse events, which involves secure handling of sensitive patient data to ensure safety and compliance. Integrating GDPR, HIPAA, and CDSCO for Comprehensive Data Protection The frameworks, each with unique regional and operational needs, aim to protect patient data. Ensure ethical conduct, and maintain trial integrity. Key overlaps include
Beyond the Prescription: How Pharmacovigilance Safeguards Your Health

When you take a medication, you expect it to heal you—- not cause unexpected harm. But even the most rigorously tested drugs can have side effects that only appear once thousands or millions of people start using them. This is where pharmacovigilance (PV) steps in —- the quiet, constant guardian ensuring medicines remain safe long after they reach the market. At CUREX, we believe that an informed patient and a proactive healthcare provider form the best defense against drug-related harm. That’s why PV isn’t just part of our operations—- it’s at the heart of our mission. What exactly is Pharmacovigilance? Pharmacovigilance is the science and practice of detecting, assessing, understanding, and preventing adverse effects or any drug-related problems. Think of it as a safety net that operates continuously, ensuring that real-world experience with a drug is closely monitored and analyzed. PV involves: Figure 1: Pharmacovigilance in CUREX “One patient’s experience can lead to safety measures that protect millions.” Why Pharmacovigilance Matters More Than Ever Bridging the Clinical Trial Gap -Clinical trials involve a limited number of participants under controlled conditions. PV extends this by tracking safety in diverse, everyday populations — elderly patients, pregnant women, people with multiple health conditions — uncovering risks that might otherwise go unnoticed. Catching the Unpredictable -Some adverse effects occur in less than 1 in 10,000 patients, or appear years after treatment. PV enables the long-term, large-scale observation needed to detect these rare events. Ensuring Generic & Biosimilar Safety -While generics match branded drugs chemically, differences in manufacturing can impact safety or efficacy. PV ensures these alternatives meet the same high safety standards. Preventing Dangerous Drug Interactions -By analyzing prescription trends, PV systems can flag hazardous combinations — like blood thinners with certain painkillers — before they cause harm. The Evolution of PV: From Paper Reports to AI Insights Pharmacovigilance has transformed from a slow, manual process into a data-driven global safety network: Figure 2: Evolution of Pharmacovigilance At CUREX, we integrate anonymized, consent-based patient data into global PV databases, accelerating detection of potential risks and enabling faster safety interventions. How Patients and Providers Play a Role Pharmacovigilance thrives on community participation: Report Side Effects promptly : even if they seem minor. Small patterns can reveal big safety issues. Provide Complete Medical History: including allergies, other medications, and health conditions, to help analysts assess risks accurately. Myth: Only severe side effects are worth reporting. Truth: Mild or common side effects can be key to spotting trends and improving dosage recommendations. CUREX’s Commitment to Excellence in Pharmacovigilance We go beyond regulatory requirements, focusing on innovation and accessibility: Figure 3: CUREX Commitment to Pharmacovigilance The Bottom Line Pharmacovigilance is more than just a regulatory checkbox >>> it’s a dynamic shield protecting public health. By turning data into actionable safety measures, PV ensures that today’s medical breakthroughs don’t become tomorrow’s public health crises. At CUREX, we don’t just deliver treatments, we safeguard trust. Stay Vigilant. Stay Safe. Have a concern about your medication? Submit a report via our Safety Portal or speak directly with your CUREX care coordinator. read also: Exploring Clinical Trials: A Comprehensive Guide to Medical Innovation



