

The pharmaceutical and biotechnology industry moves fast, but regulatory approval still remains one of the biggest barriers between a promising molecule and a commercially available therapy. Companies investing millions into drug discovery and clinical development quickly realize that scientific innovation alone is not enough. Without a well-structured regulatory affairs strategy, even highly effective products can face delays, clinical holds, Refuse-to-File (RTF) actions, or complete rejection from health authorities.
In 2026, IND submissions, NDA submissions, and global regulatory compliance expectations continue evolving toward more data standardization, electronic submissions, risk-based review processes, and structured clinical protocol frameworks. Regulatory agencies now expect stronger documentation quality, improved pharmacovigilance planning, robust clinical data management, and validated electronic systems throughout the drug development lifecycle.
For pharmaceutical companies, biotech startups, CROs, and regulatory professionals, mastering IND/NDA submissions is no longer just a compliance task. It is a strategic capability that directly affects development timelines, investor confidence, product commercialization, and long-term market success.
What Are IND and NDA Submissions?
Every drug approved in the United States follows a structured regulatory pathway, and the two most important milestones in that journey are the Investigational New Drug (IND) submission and the New Drug Application (NDA) submission. These applications form the backbone of modern regulatory affairs services and determine whether a sponsor can begin human clinical trials and later market the product commercially.
An IND submission is submitted before human clinical trials begin. Its primary purpose is to demonstrate that the investigational product is reasonably safe for testing in humans. Regulatory authorities review preclinical pharmacology, toxicology data, manufacturing information, and proposed clinical protocols before allowing the study to proceed.
An NDA submission, on the other hand, represents the complete evidence package submitted after successful clinical development. The NDA demonstrates that the drug is safe, effective, consistently manufactured, and appropriately labeled for commercial use. This submission includes extensive data from nonclinical studies, clinical trials, clinical data management systems, Chemistry Manufacturing and Controls (CMC), pharmacovigilance services, statistical analyses, and risk management documentation.
Think of the IND as obtaining permission to begin the journey, while the NDA is the final request for market authorization. One opens the clinical research door. The other determines commercial success.
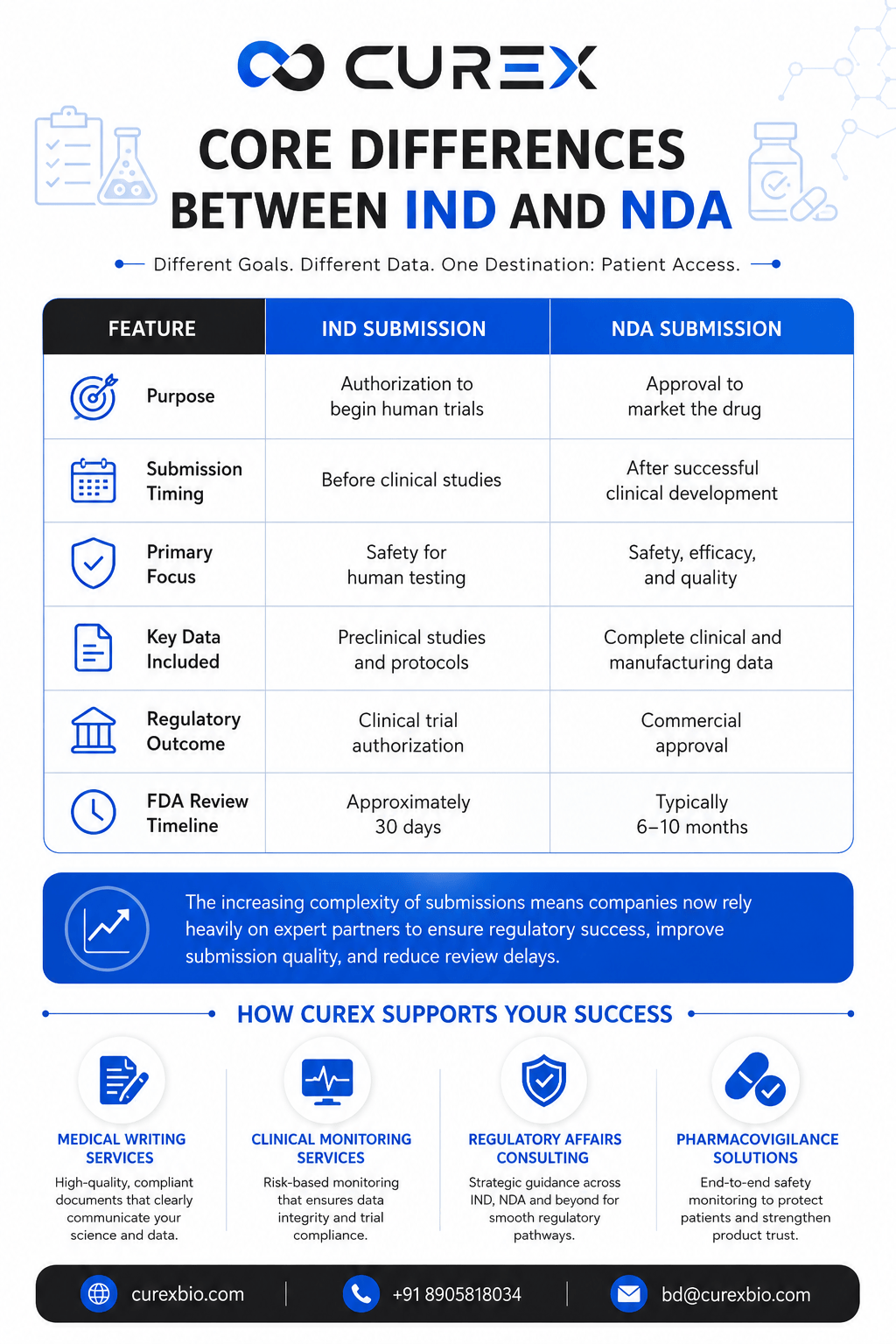
Core Differences Between IND and NDA
| Feature | IND Submission | NDA Submission |
| Purpose | Authorization to begin human trials | Approval to market the drug |
| Submission Timing | Before clinical studies | After successful clinical development |
| Primary Focus | Safety for human testing | Safety, efficacy, and quality |
| Key Data Included | Preclinical studies and protocols | Complete clinical and manufacturing data |
| Regulatory Outcome | Clinical trial authorization | Commercial approval |
| FDA Review Timeline | Approximately 30 days | Typically 6–10 months |
The increasing complexity of submissions means companies now rely heavily on medical writing services, clinical monitoring services, regulatory affairs consulting, and pharmacovigilance solutions to improve submission quality and reduce review delays.
The Modern Drug Development and Regulatory Lifecycle
Drug development is often described as a marathon disguised as a science project. A single product may require 10 to 15 years of development before approval, with billions of dollars invested across discovery, preclinical testing, clinical trials, manufacturing scale-up, and post-marketing surveillance.
The regulatory lifecycle typically includes:
- Drug discovery and candidate selection
- Preclinical testing
- IND submission
- Phase I clinical trials
- Phase II clinical trials
- Phase III clinical trials
- NDA submission
- Regulatory review and approval
- Post-marketing pharmacovigilance
Modern regulatory agencies increasingly expect sponsors to integrate compliance planning from the earliest development stages. Companies that delay regulatory affairs planning often encounter major submission deficiencies later in the process.
Preclinical Development Requirements
Before an IND submission can occur, sponsors must generate sufficient nonclinical evidence demonstrating the investigational drug is reasonably safe for human exposure.
These studies help determine:
- Potential toxicity risks
- Pharmacokinetic behavior
- Dose selection strategies
- Safety margins before first-in-human administration
The preclinical phase also establishes foundational CMC documentation, which becomes critically important later during NDA review. Manufacturing inconsistencies remain one of the leading causes of regulatory delays globally.
This is why many sponsors now integrate:
- Bioanalytical services
- PK/PD analysis
- Scientific affairs support
- Clinical quality compliance services
- Regulatory affairs consulting
early in development rather than treating regulatory documentation as a final-stage activity.
Strengthen Your IND & NDA Submission Strategy
Need support with regulatory affairs services, medical writing services, clinical data management, or pharmacovigilance services?
Clinical Trial Phases and Regulatory Expectations
Once the IND becomes active, clinical development officially begins. Clinical trials progress through multiple phases, each carrying different regulatory objectives and risk profiles.
Phase I trials primarily evaluate safety, tolerability, pharmacokinetics, and dose escalation. These studies usually involve small participant populations and emphasize patient safety monitoring.
Phase II trials focus on early efficacy signals, dose optimization, and expanded safety evaluation. This stage often determines whether development continues or stops.
Phase III trials involve larger patient populations and generate the pivotal evidence required for NDA approval. Regulatory agencies expect robust statistical analysis plans, protocol compliance, adverse event reporting, and validated clinical endpoints.
The increasing complexity of global studies means sponsors must now manage:
- Multi-country regulatory submissions
- Electronic Trial Master Files (eTMF)
- Data integrity requirements
- Centralized monitoring
- GCP compliance expectations
- Safety signal detection
- Risk-based monitoring
Regulatory agencies also increasingly evaluate operational quality systems during inspections. Sponsors are expected to demonstrate oversight of CROs, vendors, and computerized systems used throughout development.
Organizations investing in clinical monitoring services, clinical site management services, and clinical trial solutions are often better prepared for inspections and submission reviews.
Key Components of an IND Submission
Preparing a successful IND is both a scientific and strategic exercise. A poorly structured IND can lead to delays, regulatory questions, or clinical holds that significantly impact development timelines.
A complete IND submission generally includes:
- Clinical protocols
- Investigator information
- Investigator brochures
- Preclinical pharmacology and toxicology data
- Manufacturing and CMC information
- Previous human experience data if available
The challenge is not simply collecting these documents. The challenge lies in presenting them clearly, accurately, and strategically.
Nonclinical Data Requirements
Nonclinical studies form the scientific foundation supporting human exposure. Regulatory reviewers carefully assess whether the available toxicology and pharmacology evidence adequately supports the proposed clinical trial.
Sponsors must provide detailed information on:
- Acute and chronic toxicity
- Genotoxicity
- Reproductive toxicity
- Carcinogenicity when applicable
- Pharmacokinetics
- Pharmacodynamics
- Dose-response relationships
Poorly interpreted toxicology findings can trigger extensive regulatory questions. This is where experienced medical writing services and scientific affairs consulting become essential.
Regulatory reviewers want concise, scientifically defensible summaries—not overwhelming volumes of disconnected raw data.
Simplify Complex Regulatory Submissions
From clinical monitoring services to regulatory affairs consulting and safety pharmacovigilance, CurexBio helps sponsors improve submission readiness and compliance efficiency.
Clinical Protocol and Investigator Documentation
Clinical protocols are among the most heavily scrutinized parts of an IND submission. Regulatory agencies expect protocols to include clear objectives, safety monitoring procedures, endpoint definitions, and statistical methodologies.
Modern protocol development now focuses on:
- Clear endpoint definitions
- Statistical transparency
- Risk mitigation strategies
- Patient safety protections
- Electronic protocol standardization
- Operational feasibility
The growing adoption of structured electronic protocol standards is improving:
- Regulatory review consistency
- Cross-functional collaboration
- Clinical operations efficiency
- Data interoperability
- Inspection readiness
Companies that adopt standardized protocol development approaches early often improve operational efficiency during global clinical programs.
NDA Submission Strategy and Approval Pathway
The NDA is often described as the “final exam” of drug development. Every clinical observation, manufacturing process, analytical method, and safety assessment eventually converges into this massive regulatory submission.
Modern NDAs may contain hundreds of thousands of pages organized into electronic Common Technical Document (eCTD) format. Regulatory reviewers evaluate every aspect of the product lifecycle, including:
- Clinical efficacy
- Safety profile
- Risk-benefit assessment
- Labeling accuracy
- Manufacturing consistency
- Pharmacovigilance planning
- Statistical validity
- Data integrity compliance
A successful NDA requires far more than strong clinical trial results. Many products fail because of operational weaknesses, manufacturing deficiencies, or inconsistent regulatory documentation.
Common Technical Document (CTD) Structure
Most global submissions now follow the Common Technical Document (CTD) format.
| CTD Module | Content |
| Module 1 | Regional administrative information |
| Module 2 | Summaries and overviews |
| Module 3 | Quality and CMC data |
| Module 4 | Nonclinical study reports |
| Module 5 | Clinical study reports |
Electronic submissions are now considered standard practice, with increasing emphasis on:
- eCTD compliance
- Metadata accuracy
- Structured datasets
- Traceability
- Electronic review compatibility
The regulatory landscape is rapidly evolving toward digital-first submissions, automated validation checks, and structured content management systems.
Emerging Regulatory Trends in 2026
Regulatory affairs services are changing faster than many organizations anticipated. Artificial intelligence, decentralized clinical trials, structured electronic protocols, and advanced analytics are reshaping both development and regulatory review processes.
Several important trends are defining 2026:
- Greater emphasis on structured electronic submissions
- Increased focus on data integrity and ALCOA+ principles
- Expansion of decentralized trial oversight
- More flexible CMC expectations for advanced therapies
- Increased use of innovative statistical methodologies
- Stronger pharmacovigilance signal detection expectations
Organizations investing in modern clinical trial solutions, clinical data management systems, and regulatory intelligence services are better positioned to adapt to these evolving expectations.
ICH M11 and Structured Clinical Protocols
The ICH M11 initiative represents one of the most important protocol harmonization efforts in recent years. Historically, protocol formats varied widely between sponsors, leading to inefficiencies, inconsistent review processes, and operational confusion.
The M11 framework aims to create:
- Harmonized protocol templates
- Standardized content organization
- Electronic interoperability
- Improved review efficiency
- Better protocol traceability
Regulatory professionals increasingly expect M11 adoption to become a major industry standard in coming years. Sponsors preparing future global programs should already begin evaluating alignment strategies.
Common IND/NDA Submission Challenges
Even experienced organizations encounter submission difficulties. Regulatory failures rarely stem from one catastrophic issue. Instead, they usually emerge from cumulative operational weaknesses.
Some of the most common challenges include:
| Regulatory Challenge | Impact |
| Incomplete documentation | Submission delays |
| Poor protocol design | Regulatory questions and amendments |
| Inconsistent CMC data | Approval risk |
| Weak pharmacovigilance planning | Safety concerns |
| Data integrity gaps | Inspection findings |
| Poor eCTD formatting | Technical rejection |
| Insufficient statistical justification | Review delays |
Many biotech startups underestimate the complexity of regulatory operations until late-stage development. By then, correcting deficiencies becomes expensive and time-consuming.
That is why sponsors increasingly outsource specialized activities such as:
- Regulatory affairs services
- Medical writing services
- Clinical data management
- Pharmacovigilance services
- Clinical monitoring services
- Biostatistics services
- Safety database management
The goal is no longer just regulatory compliance. The goal is submission optimization.
How Regulatory Affairs Services Improve Submission Success
Modern regulatory affairs consulting teams function as strategic partners across the entire development lifecycle. They help organizations interpret evolving guidance, prepare compliant documentation, manage authority interactions, and reduce operational risks.
An experienced regulatory affairs partner can support:
- IND strategy planning
- Regulatory meeting preparation
- Gap analysis
- eCTD publishing
- Labeling review
- CMC documentation
- Submission lifecycle management
- Global registration strategy
Integrated service models are becoming increasingly valuable because they connect regulatory expertise with operational execution.
For example, combining:
- Clinical data management services
- Medical writing services
- Pharmacovigilance services
- Regulatory affairs consulting
- Scientific affairs support
- Clinical monitoring services
within a coordinated framework significantly improves submission consistency and communication efficiency.
The future of regulatory affairs is not just compliance-driven. It is data-driven, technology-enabled, and strategically integrated across development functions.
Conclusion
IND and NDA submissions represent far more than regulatory paperwork. They are strategic milestones that determine whether promising therapies successfully reach patients and global markets.
As regulatory agencies continue modernizing review processes, sponsors must adapt to increasing expectations around structured submissions, electronic interoperability, data integrity, patient safety, and operational quality systems.
Organizations that invest early in strong regulatory affairs services, integrated quality systems, expert medical writing services, advanced pharmacovigilance services, and submission readiness strategies will be better positioned to accelerate approvals and minimize costly delays.
Successful regulatory execution no longer depends on isolated submission preparation. It depends on building connected, compliant, and scalable development ecosystems from the very beginning.
FAQs
What is the primary purpose of an IND submission?
An IND submission allows sponsors to obtain authorization to begin clinical testing of an investigational drug in humans. The submission demonstrates that the product is reasonably safe for early-stage clinical trials.
What is included in an NDA submission?
An NDA includes complete clinical, nonclinical, manufacturing, labeling, pharmacovigilance, and statistical data demonstrating that the drug is safe and effective for commercial use.
Why are regulatory affairs services important?
Regulatory affairs services help organizations navigate evolving compliance requirements, improve submission quality, manage authority interactions, and reduce approval delays across global development programs.
How do pharmacovigilance services support NDA submissions?
Pharmacovigilance services help sponsors monitor adverse events, manage safety databases, conduct signal detection, and maintain ongoing regulatory safety compliance.
Why are medical writing services important during IND/NDA submissions?
Medical writing services help create clear, scientifically accurate, and submission-ready documents including clinical study reports, protocols, investigator brochures, and regulatory summaries.



