
Regulatory Affairs Pre-Submission Checklist: Building Bulletproof Protocols for Regulatory Success

If you work in the biotech or pharmaceutical sectors, you understand that an inadequately structured protocol can significantly hinder a regulatory submission, leading to substantial financial losses and time delays in market entry. At CurexBio, we have observed instances where exceptional scientific efforts falter due to preventable protocol errors in regulatory affairs during the submission phase. To address this issue, we are providing a detailed, actionable checklist grounded in experience for constructing high-quality, submission-ready protocols. Adhering to these guidelines will enhance your dossier, regulatory affairs submission, fulfill reviewer expectations, and maintain your development timeline. Protocol Quality Matters More Than Ever-Why? Regulatory agencies such as the FDA and EMA are not only examining data but also the processes behind it. An unclear or poorly designed protocol can indicate larger quality issues, leading to an increase in refusal-to-file letters, clinical holds, and extensive information inquiries tied to protocol flaws. Here, CurexBio focuses on protocol optimization and regulatory preparedness, providing guidance for early- to mid-stage biotechnology companies to meet these essential criteria effectively. Actionable Steps for Pre-Submission Protocol Checklist in Regulatory Affairs Figure 1: Steps for Pre-submission Protocol Checklist Phase 1: Elementary Alignment (Before Drafting Begins) Specify Primary & Secondary Objectives Clearly Ensure each objective is SMART: Specific, Measurable, Achievable, Relevant, Time-bound. Vague objectives lead to ambiguous endpoints—a major red flag for regulators. Conduct a Robust Literature & Competitor Review Understand precedents, standard-of-care expectations, and relevant guidance documents (FDA, ICH, EMA). Cite appropriately. Engage Early with Regulatory Consultants or Internal QA A preliminary gap analysis can save countless revisions later. Curex Bio often performs pre-protocol regulatory assessments to identify alignment issues before the first draft. Map Out All Required Protocol Sections Use agency templates (e.g., FDA Form 1572 supporting protocols, ICH E6 (R2) structure) as an examples. Phase 2: Drafting with Precision Write with Unambiguous Language Avoid “as needed,” “where appropriate,” or “regularly.” Define all terms. Use consistent terminology. Detail Inclusion/Exclusion Criteria with Justification Explain why each criterion is scientifically and ethically justified. Overly restrictive criteria can raise enrollment concerns; overly broad criteria threaten data integrity. Define Endpoints with Operational Clarity Specify each endpoints and its measurement procedure with personal details, timepoints, methodologies/tools, equipment, and scoring systems. Describe Detail Statistical Methods Include sample size justification, power calculations, handling of missing data, interim analysis plans, and stopping rules. Address Risk-Based Monitoring (ICH E6 R2) Outline centralized monitoring processes, key risk indicators, and plans for targeted on-site visits. Show you’re prioritizing data-critical elements. Incorporate Patient Safety & Ethical Protections Detail safety monitoring, reporting timelines for SAEs, DSMB charters, and patient informed consent processes. Phase 3: Internal Cross-Functional Review Conduct a Formal Protocol Review Meeting Include clinical operations, biostatistics, data management, regulatory affairs, pharmacovigilance, and medical monitors. Document feedback and resolutions. Verify Consistency Across All Documents Cross-check the protocol with the clinical study report template, statistical analysis plan, informed consent form, and case report forms. Inconsistencies invite queries. Perform a Mock Agency Review Have someone not involved in drafting simulate a regulatory review. CurexBio offers protocol challenge sessions designed to stress-test protocols against typical reviewer comments. Check Formatting & Referencing Ensure version control, clear dating, complete references, and compliance with electronic submission standards. Phase 4: Finalization & Submission Readiness QA Sign-Off Confirm all checklist items are completed and documented. Compile a Protocol Synopsis or Summary Many agencies appreciate a clear, concise overview (1–2 pages) for quick assessment. Update the Trial Registry Entry Ensure ClinicalTrials.gov or other registry entries match the final protocol exactly. Archive All Supporting Documentation Include literature, meeting minutes, statistical justifications, and correspondence with experts or regulators. Curex Bio Supports Your Protocol Success-How? We know that small and mid-sized teams often lack dedicated protocol authorship experts. That’s where we add value: End-to-End Protocol Development involves creating submission-ready documents that meet agency expectations, ensuring a comprehensive process from initial concept to final submission. Gap Analysis and Pre-Submission Review involve identifying weaknesses in processes or submissions prior to regulatory scrutiny, thereby enabling organizations to address potential issues proactively. Modular Protocol Templates are customizable, ICH-compliant tools that enhance the efficiency of drafting while ensuring quality standards are met. Training & Workshops: Equip your team with optimal approaches in protocol design and regulatory strategy. Our clients confidently progress toward submission by avoiding pitfalls such as ambiguous endpoints, inconsistent statistical plans, and inadequate risk monitoring—common challenges encountered during regulatory affairs submissions. A high-quality protocol serves as a strategic document that reflects a company’s scientific integrity and operational effectiveness, transforming protocol development within regulatory affairs from a compliance challenge into a competitive asset through a systematic, checklist-based approach. Need expert support? CurexBio offers expert support for biotech and pharmaceutical firms, providing specialized regulatory affairs strategy and documentation services to facilitate quicker and more efficient approvals through high-quality submissions. Contact Us today for a free preliminary assessment to ensure your protocol submission is ready for regulatory review
Navigating the Future of Clinical Trials: ICH E20 & The Power of Adaptive Designs
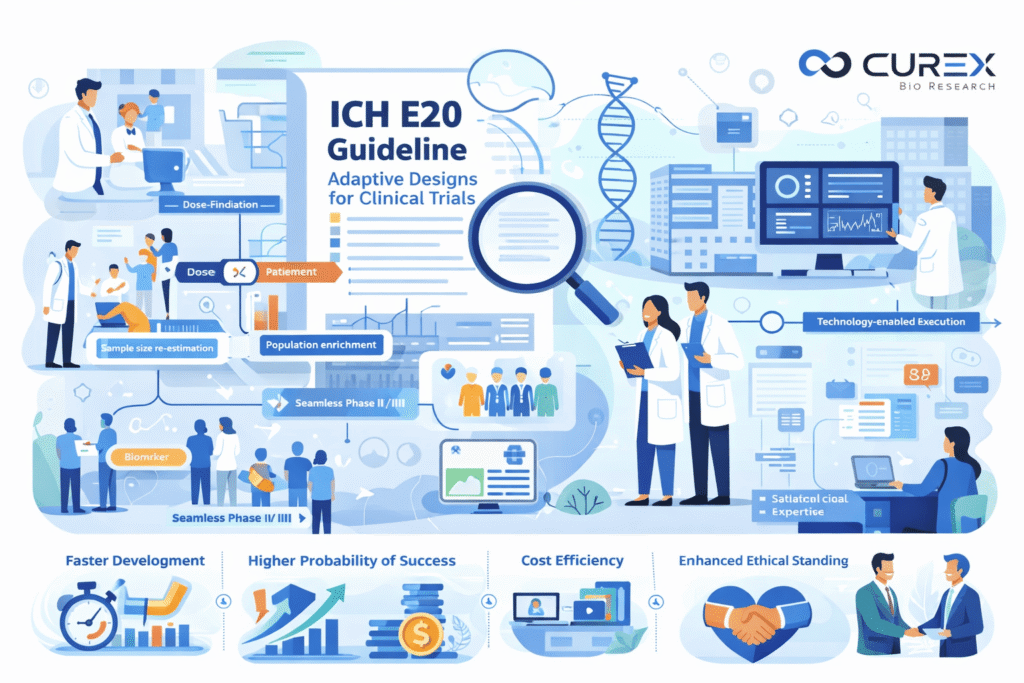
The clinical development landscape is experiencing significant changes, moving away from traditional, linear, and fixed Clinical Trials designs. These conventional methods are increasingly seen as rigid, inefficient, and not fully capable of addressing the complexities inherent in modern medicine. In response, adaptive clinical trial designs have emerged as a transformative strategy, enabling planned and data-driven adjustments throughout the trial process. To provide a comprehensive framework for this innovative approach, the International Council for Harmonization (ICH) introduced the pivotal ICH E20 guideline in 2023. This guideline aims to establish global clarity and standards for adaptive trials, paving the way for more flexible and responsive clinical research methods. In this context, we aims to clarify the ICH E20 guideline, highlight the advantages of adaptive designs, and CurexBio is adeptly prepared to navigate these advanced development pathways to aid in the progress of breakthrough therapies. What is ICH E20 and Why Does It Matter? ICH E20, titled “Adaptive Designs for Clinical Trials,” is the inaugural internationally harmonized guideline focused on the planning and execution of adaptive trials. Its publication represents a significant milestone, reflecting global regulatory acceptance and offering a comprehensive framework for sponsors involved in such clinical studies. Figure 1: Core Mission of ICH E20 Common Types of Adaptive Designs include: Figure 2: Types of Adaptive clinical trials Dose-Finding Designs: Efficiently identify the optimal therapeutic dose by allocating more patients to promising dose levels and away from toxic or ineffective ones. Adaptive Randomization: Adjust the randomization probability to favor the treatment arm showing better interim results, offering ethical benefits to participants. Sample Size Re-Estimation: Recalculate the required number of patients at an interim analysis based on observed variability or treatment effect, ensuring the trial is neither under- nor over-powered. Population Enrichment: Refine the target patient population during the trial to focus on those most likely to respond (e.g., based on a biomarker). Seamless Phase II/III Trials: Combine proof-of-concept and confirmatory stages into a single, continuous trial, saving significant time and resources. Advantages of Adaptive clinical trials: Faster Development: Get effective therapies to patients sooner. Higher Probability of Success: Make informed decisions mid-trial to focus resources on winning strategies. Cost Efficiency: Reduce the number of patients exposed to ineffective treatments and optimize resource allocation. Enhanced Ethical Standing: Treat trial participants more intelligently and compassionately. The Challenge: Complexity and Regulatory Scrutiny The power of adaptive designs comes with complexity. Their success hinges on: Meticulous Pre-Planning: Every potential adaptation must be exhaustively detailed in the protocol and statistical analysis plan *before the trial begins*. Advanced Statistical Expertise: Requiring sophisticated simulation and modeling to control Type I error and avoid operational bias. Robust Operational Infrastructure: Dynamic randomization, real-time data capture, and stringent interim analysis processes with firewalls. Proactive Regulatory Engagement: Clear communication and alignment with health authorities is non-negotiable. Curex Bio Empowers Your Adaptive Design Strategy A poorly planned adaptive trial may result in invalid outcomes, regulatory non-acceptance, and squandered investments. In such scenarios, having a specialized partner like CurexBio proves to be essential. At CurexBio, we focus on providing a comprehensive strategic partnership aimed at maximizing the effectiveness of ICH E20 and adaptive designs within clinical programs. Our integrated approach enhances collaboration and supports the overall success of clinical initiatives. Strategic Design & Simulation: Our team of biostatisticians and clinical scientists collaborates with clients to assess the appropriateness of adaptive design for their objectives. We explore various design options, quantify essential operating characteristics such as power and sample size distributions, and develop a comprehensive, pre-specified adaptation plan that meets regulatory standards and instills confidence. Regulatory Pathway Mastery: We develop thorough documentation in accordance with ICH E20 standards, which encompasses a detailed adaptive design charter. Our regulatory affairs team is instrumental in preparing for and facilitating essential interactions with the FDA, EMA, and other regulatory agencies, ensuring that all processes are aligned right from the beginning. Technology-Enabled Execution: We implement and manage integrated technology platforms that facilitate real-time data collection, centralized monitoring, and dynamic randomization processes. These measures are essential for ensuring data integrity and preventing operational bias during critical adaptation points. Operational Excellence & Risk Mitigation: Our clinical operations team specializes in the distinct requirements of adaptive trials, creating comprehensive playbooks for a variety of adaptation scenarios. We take a proactive approach in identifying and mitigating risks, which allows for the seamless execution of complex trials across multiple global sites. Why Partner with Curex Bio? Deep Knowledge Our team includes veterans who have successfully designed, defended, and executed adaptive trials across therapeutic areas. Flexible Partnership: We integrate seamlessly with your team, offering flexibility and transparency. Focus on Your Success: We are committed to making your trial a model of efficiency and scientific integrity, accelerating the path to approval. Ready to design a smarter, faster clinical trial? Contact CurexBio today to explore how our adaptive design services can de-risk and accelerate your path to market.
Trial Efficiency vs. Trial Speed: How Expert Clinical Development Services Truly Accelerate Timelines

In the competitive arena of clinical development, the urgency to expedite the introduction of new therapies is paramount. Sponsors face significant pressure to accelerate timelines, control rising costs, and meet the expectations of investors. As a result, the prevailing mantra within this context is often a demand for increased speed in trial execution—something that Expert Clinical Development Services can help achieve more strategically and sustainably. However, this immediacy can lead to a pervasive and potentially detrimental misunderstanding: conflating “trial speed” with “trial efficiency”. It is crucial to recognize that while these two concepts are interconnected, they do not embody the same principle. The ability to distinguish between mere speed and strategic efficiency can be the determining factor that separates a well-planned, accelerated clinical program from one that operates in a chaotic, hasty manner. This confusion arises from the high stakes and pressures inherent in the clinical trial landscape. To navigate these complexities successfully, aligning with Expert Clinical Development Services emerges as a pivotal strategy. Such partnerships can facilitate the enhancement of genuine efficiency, which results in meaningful speed that is both measurable and sustainable, rather than simply a frantic rush towards completion. Defining the Two Concepts Trial Speed: Defined as a raw, output-focused metric that quantitatively measures the rate of progress of a study through its critical milestones. Key performance metrics: Include the patient enrolment rate, the duration from First Patient First Visit (FPFV) to Last Patient Last Visit (LPLV), the timeline for site activation, and the time taken to achieve database lock. Trial efficiency: Is a resource-centered approach aimed at achieving milestones while optimizing the use of time, money, and effort? This philosophy emphasizes the importance of executing processes correctly on the first attempt, thereby minimizing waste and enhancing operational efficiency. Key metrics used to gauge trial efficiency include cost per enrolled patient, the rate of protocol amendments, query resolution times, accuracy in data entry, and the performance and engagement levels of sites involved in the trials. Global regulatory bodies emphasize that protocol quality and operational efficiency directly impact trial timelines. Refer to ICH E6 R2 guidance for detailed expectations: https://ich.org/page/efficacy-guidelines. Why Sponsors Consistently Mix Them Up The conflation is understandable and stems from several common pressures: The Urgency of Patient Need: In therapeutic areas characterized by significant unmet medical needs, there exists a strong moral imperative to expedite action. This urgency, however, can sometimes diminish the importance of careful strategic planning that is crucial for achieving efficient progress. Investor & Market Pressure: Quarterly reports and competitive landscapes necessitate evident advancements in metrics. The phrase “We activated 50 sites in one month!” conveys a sense of immediacy and impressive progress, in contrast to “We improved our patient screening process, reducing screen failures by 15%.” While the latter indicates a significant efficiency enhancement, the former creates an impression of quicker results. Distinct Perspectives: Different departments within an organization prioritize distinct metrics: Finance emphasizes cost management, clinical operations focuses on enrolment figures, and leadership tracks the calendar. This segmentation can lead to conflicting objectives, where an exclusive focus on one metric, such as speed, may inadvertently undermine another important metric, like efficiency. Therefore, it’s crucial to maintain a comprehensive perspective that harmonizes all metrics across departments. Misinterpreting “Fast” Actions: A common mistake in clinical research is attempting to solve problems quickly by allocating more resources, such as hiring additional Clinical Research Associates (CRAs), offering high patient recruitment bonuses, or hastily finalizing protocols. While this approach may yield immediate results, it can result in long-term inefficiencies, including budget overruns, data inconsistencies, and expensive amendments. The High Cost of Confusing Speed for Efficiency Prioritizing raw speed over thoughtful efficiency leads to predictable and expensive consequences: Protocol Amendments: A rushed protocol can lead to significant flaws, resulting in amendments that have the potential to delay a study by several months and incur costs amounting to millions of dollars. Poor Site Selection & Engagement: Activating clinical trial sites quickly is important, but prioritizing the right sites that can effectively recruit participants and perform tasks is crucial for efficiency. Inefficiently chosen sites can hinder progress and negatively impact project timelines. Data Quality Issues: Rushing data entry and management can lead to numerous queries and complications later, turning the data clean-up and database lock phase into a challenging and problematic situation that nullifies any initial advantages gained from the speed of data processing. Burnout and High Turnover: Frantic and poorly managed timelines in clinical settings contribute to burnout among clinical teams. This burnout results in high turnover rates among Contract Research Organization (CRO) and sponsor staff, culminating in a significant loss of valuable institutional knowledge. The need for a shift in perspective regarding clinical trials. Rather than focusing solely on speed, sponsors should concentrate on enhancing the efficiency of every process involved. This approach is highlighted as the fundamental value of Expert Clinical Development Services. It asserts that an experienced partner’s role extends beyond mere task execution; that CurexBio can be helpful in embedding efficiency into the trial’s structure from the outset. Protocol Optimization: This is an essential managing efficiency, where experts utilize feasibility data, predictive analytics, and operational input to create protocols that are scientifically validated and practically implementable, significantly lowering amendment risks. Intelligent Site Selection: Experts utilizes data-driven insights not just to compile a list of sites, but to pinpoint and engage those with established access to suitable patient populations and a track record of delivering high-quality performance. Proactive Risk Management: Risk-based monitoring (RBM) and centralized data surveillance are employed to proactively identify and mitigate issues before they can affect timelines or data integrity, rather than merely reacting to problems as they arise. Technology Integration: Leveraging eClinical technologies enhances data flow, improves site experience, and enables real-time visibility for expedited decision-making. Strategic Patient Engagement: Clinical trial expert create recruitment plans that utilize predictive modeling and digital outreach to effectively target the appropriate patients. Don’t Just Accelerate—Optimize with CurexBio’s Expert Clinical Development Services In this metaphorical analogy, the process of bringing a product
The Unseen Guardian: How Biostatistics Protects Your Clinical Trial Data

In clinical research, data integrity serves as the cornerstone for conclusions, regulatory submissions, and patient safety. While routine checks and validations provide a basic level of defense, more complex risks can exist within seemingly clean data. To address these hidden threats, statistical monitoring enhanced by biostatistics elevates the role of Clinical Data Managers (CDMs), shifting them from mere quality checkers to proactive risk detectors. For data managers involved with research sites, sponsors, and Contract Research Organizations (CROs), it is imperative to transition from manual verification to statistically-driven surveillance to uphold trial integrity and efficiency. The Statistical Toolkit: Z-Scores, Outliers, and Trends At its core, statistical monitoring uses the data itself to flag anomalies that human reviewers might miss. Here’s how key tools work: Z-Scores A Z-score measures the distance in standard deviations of a data point from its group mean, serving as a practical alarm system in CDM, rather than an academic tool. Practical Application: Calculating Z-scores for systolic blood pressure readings at Site A allows for immediate identification of anomalously high or low values. A concentration of extreme Z-scores may suggest issues such as protocol deviations, measurement errors, or gaps in training. Outlier Detection Outliers are data points that deviate from expected variation, and statistical models are used to differentiate between natural variability and problematic anomalies. Practical Application: Statistical outlier detection can identify clinically significant shifts in a patient’s creatinine value, which may indicate adverse events or data entry errors. This approach compares the value against other patients in the same treatment arm or the patient’s own baseline, rather than relying solely on the lab’s normal range. Trend Analysis This is where data managers predict and prevent issues by examining trends that analyze data over time or across groups to identify systematic patterns. Practical Application: Temporal Trends: There is a concerning trend of steadily increasing missing pages in electronic Case Report Forms (eCRFs) at a site, indicating a potential future data quality crisis. Cross-Site Trends: Patients at a site reporting zero pain scores may indicate a bias in assessment, known as “digit preference.” Treatment Arm Trends: Early detection of imbalances in baseline characteristics during enrollment is crucial for enabling corrective actions. Implementing Statistical Monitoring Inside the CDM Process: A Biostatistics-Driven Approach Targeted Source Data Verification (SDV): Move from 100% SDV to risk-based monitoring, focusing resources on sites and variables with statistical flags. Earlier Issue Resolution: Detect systemic problems weeks or months before traditional cleaning cycles would catch them. Enhanced Patient Safety: Identify potential safety signals (through outlier lab trends) buried within accumulating data. Regulatory Confidence: Demonstrate to regulators a proactive, sophisticated, and quantitative approach to data quality oversight. How CurexBio Empowers Your Data Management Team At CurexBio, we connect biostatistics theory with practical clinical data management. Our approach goes beyond providing tools; we embed statistical intelligence directly into your workflow to enhance efficiency and effectiveness. Our Statistical Monitoring & Risk Detection Service provides: Embedded Analytics: Automated and scheduled reports provide data managers with direct access to Z-score, outlier, and trend analyses. These reports are customized to focus on the specific endpoints and risks pertinent to each study, enhancing the monitoring and management of data throughout the research process. Risk Scorecards: Dynamic visual scorecards assess and rank each site based on statistical risk metrics, facilitating prioritized actions to address identified risks effectively. Expert Partnership: Our biostatisticians and data management experts collaborate with your team to analyze data flags, differentiating between genuine risks and irrelevant data. They also assist in formulating actionable corrective and preventive actions (CAPAs) based on the analysis. Training & Upskilling: We provide your Clinical Trial Data Management team with the necessary knowledge to comprehend and utilize statistical techniques, thereby fostering enduring in-house capabilities. In contemporary clinical trials, data managers play a crucial role as the protectors of data integrity and accuracy. CurexBio offers biostatistics, and statistical monitoring solutions, equipping these data managers with the tools necessary to enhance their oversight. By applying a proactive approach to risk management rather than simply reacting to data discrepancies, CurexBio enables these professionals to mitigate potential issues proactively, ensuring the reliability of the trial outcomes. Interested parties are encouraged to reach out to CurexBio for a discussion on how their customized biostatistics services can streamline and fortify the clinical development process, effectively reducing risks associated with data management.
From Detection to Closure: How Clinical Data Management Services Streamline the Query Life Cycle
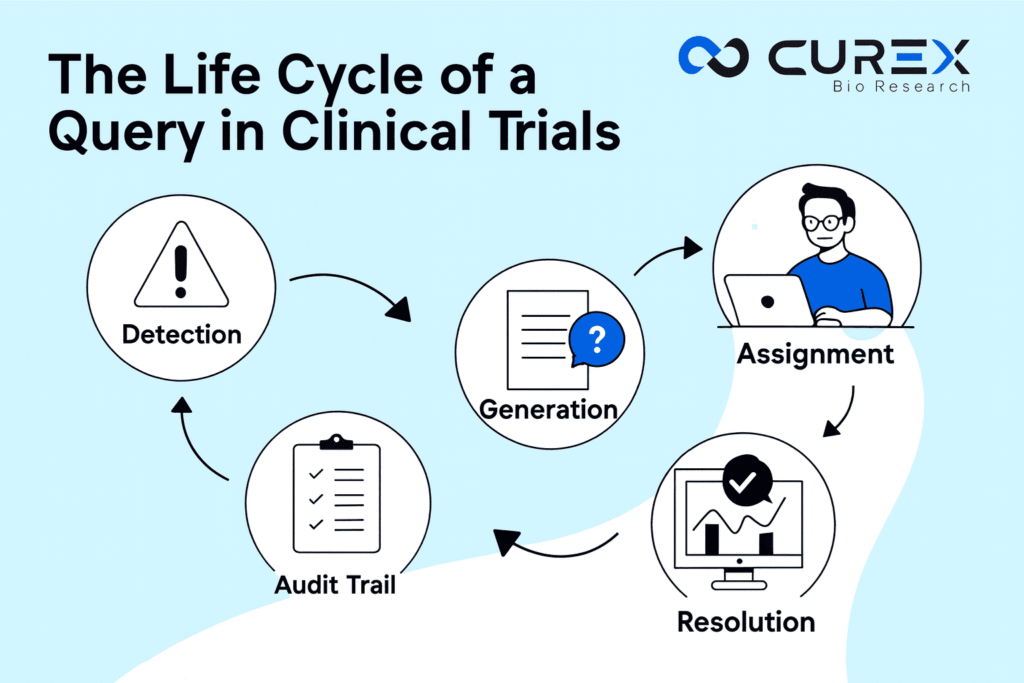
In clinical trials, data integrity is crucial, yet even meticulously designed studies can encounter issues due to inconsistencies or gaps in the collected data. Query management plays a critical role in addressing these problems, serving as a mechanism to catch and rectify errors before the final analysis. This blog explores the complete life cycle of a query—from identification to resolution—while highlighting common areas where time is lost, complicating what could be a straightforward correction. For those involved in trial operations or delivering end to end clinical data management services, grasping this cycle is essential for optimizing workflows and enhancing data quality. Why Query Management Matters in Clinical Trials At its core, query management serves to enhance communication among data teams, monitors, and site staff, facilitating the identification and rectification of erroneous data. Queries act as focused inquiries that expose missing information, irregularities, or significant errors, which are crucial in ensuring the reliability of clinical trials. Neglecting query management can lead to unreliable results, risking regulatory compliance under ICH E6(R3) Good Clinical Practice guidelines. Essentially, queries are integral to maintaining data integrity, completeness, and credibility, playing a vital role in overall data cleaning initiatives that help uphold trial progress and compliance. However, the management of such queries requires careful execution; ineffective handling can lead to increased costs, resource strain, and delays in timelines. As clinical trials become increasingly complex with the integration of electronic data capture (EDC) systems, proficiency in query management is essential for anyone involved in comprehensive clinical data management services. Breaking Down the Query Life Cycle The journey of a query isn’t random—it’s a structured workflow with distinct phases. Here’s how it typically unfolds, reimagined from standard practices in the field: Detection: Spotting the Red Flags Everything begins with the identification of issues in data management, which may include mismatched dates, out-of-range laboratory values, or empty fields requiring attention. This detection is facilitated through two primary methods: automated checks integrated within Electronic Data Capture (EDC) systems that alert users to problems immediately during data entry, and manual reviews conducted by data managers, Clinical Research Associates (CRAs), or Clinical Trial Managers (CTMs). The overarching objective is to identify discrepancies early in the process to prevent more significant complications in the future. Automated checks must be finely tuned during study setup to avoid missing subtle issues, which can lead to increased manual troubleshooting. Inefficient detection may overwhelm teams with queries and slow down progress. Generation: Crafting the Right Question Once a problem is flagged, it’s time to create the query itself. Effective communication requires concise and precise messages that clearly identify issues and specify necessary actions. Vague inquiries lead to confusion and extended clarification processes, causing significant time loss due to the “ping-pong” effect of incomplete responses. Assignment: Directing It to the Right Hands In clinical trials, queries are assigned to the relevant personnel, typically site staff, investigators, or coordinators, but may also involve coders or monitors for specific concerns. This allocation often occurs digitally in EDC platforms. However, delays can arise from misrouting or unclear responsibilities, exacerbated by time zone differences and language barriers in global trials, leading to lengthy waiting periods if assignments end up in incorrect inboxes. Monitoring: Keeping Tabs on Progress Queries require proactive management and oversight, typically by data managers or Clinical Trial Managers (CTMs), who utilize dashboards to monitor aging queries and encourage timely responses. Inadequate monitoring tools can result in unresolved queries, with industry data showing average resolution times between 23 to 52 days, and in some cases, weeks or months. This phase often intensifies the “ping-pong” effect, where incomplete responses lead to reassignments and prolonged follow-ups. Resolution: Closing the Loop When a site updates data or provides explanations, the query owner reviews the response. If acceptable, the query closes; if not, further revisions are required. Multi-round clarifications frequently occur, particularly with endpoint data. A study of a cardiovascular trial revealed that 21% of queries necessitated multiple submissions, leading to prolonged closure and potential database lock delays. Strong clinical data management services help minimize these repeated cycles by improving clarity and consistency in query handling. Audit Trail: Documenting It All for Posterity No query life cycle is complete without a solid record, as the audit trail captures details such as who raised a query, the timing, actions taken, and resolutions. This documentation is essential for traceability in Good Clinical Practice (GCP) and identifying patterns like recurring errors. Insufficient documentation can lead to audits or inspections, resulting in retroactive fixes that interrupt progress. However, thorough documentation can highlight trends and help prevent future issues. Queries in clinical trials often bounce between teams, causing delays and inefficiencies. With 0.14–0.4 queries per CRF page, mid-sized trials can generate thousands, costing up to $200 each, predominantly yielding no changes. This creates significant pressure on sites managing these queries alongside patient care, while sponsors and CROs focus on compliance. Effective clinical data management services can significantly reduce these inefficiencies by optimizing processes and minimizing unnecessary queries. Key Challenges: Redundant or Irrelevant Queries: Over-configured automated checks spam sites with low-impact flags. Communication Gaps: Unclear phrasing or lack of context leads to incomplete responses. Resource Strain: High volumes near database lock force overtime, inflating budgets. Timeline Delays: Slow resolutions postpone analyses and submissions, potentially costing thousands per day. In one Phase I study review, only 28-40% of queries fixed data, underscoring the inefficiency. Best Practices to Speed Things Up Adopt these strategies to overcome delays: Standardize Everything: From data entry guidelines to query templates, consistency cuts errors upfront. Prioritize Smartly: Triage queries by impact—focus on critical endpoints first. Train and Collaborate: Cross-team sessions align everyone, reducing misunderstandings. Leverage Metrics: Track turnaround times, volumes, and rates to refine processes and strengthen overall clinical data management services outcomes. The future of technology highlights the role of AI, NLP, and predictive analytics in automating detection and generation, enabling real-time monitoring and decentralized trials that promise quicker resolutions. This shift emphasizes proactive prevention over
Why Modern Clinical Trials Depend on Advanced Clinical Data Management Systems

In the high-stakes arena of drug development, data has emerged as a vital asset. The extensive process of moving from a potentially effective molecule to an approved therapeutic solution involves the accumulation and analysis of vast amounts of data, which includes patient vital signs, laboratory results, imaging reports, and electronic patient-reported outcomes. The necessity for managing this extensive array of information has shifted from being a mere administrative function to a fundamental component integral to the success of clinical trials. In this context, a Clinical Data Management System (CDMS) evolves into an essential strategic resource, highlighting its critical role beyond simple support, as it becomes central to navigating the complexities of clinical research. The Data Deluge: Why Traditional Spreadsheets Are No Longer Enough Modern clinical trials demand sophisticated clinical data management systems due to their complexity, which includes diverse data sources, multiple stakeholders, and a strong focus on data integrity and compliance. This evolution requires trial sponsors and sites to adopt comprehensive methods rather than relying on scattered spreadsheets and paper case report forms. Decentralized: Using wearables and digital health technologies to collect data from patients at home. Global: Involving multiple sites across different countries with varying regulatory standards. Adaptive: Requiring real-time data analysis to make protocol adjustments on the fly. Manual data management is inefficient, subject to human error, and can cause major delays. Incorrect data entry or unresolved queries can jeopardize patient safety, disrupt timelines, and incur substantial costs. Modern Clinical Data Management System A sophisticated CDMS is more than just a database. It’s an integrated ecosystem designed to ensure data integrity from the moment it’s created until it’s locked for analysis. Key capabilities include: Uncompromising Data Integrity and Quality Advanced CDMS platforms implement automated edit checks and validation rules during data entry, which quickly identifies discrepancies and minimizes manual queries. This results in cleaner datasets, leading to more powerful and reliable statistical analysis. Accelerated Trial Timelines A CDMS enhances the data lifecycle through electronic data capture (EDC), remote monitoring, and automated workflows, allowing site staff to input data swiftly, enabling real-time sponsor reviews, and efficiently resolving queries, thereby reducing development time significantly. Enhanced Regulatory Compliance A top-tier CDMS is designed to comply with regulatory standards such as FDA 21 CFR Part 11 and GDPR. It offers comprehensive audit trails, user access controls, and electronic signatures, ensuring a reliable record of actions vital for regulatory submissions and audits. Real-Time Visibility and Risk-Based Monitoring Modern systems provide dynamic dashboards and analytics, offering sponsors real-time visibility into trial progress. This facilitates Risk-Based Monitoring (RBM), enabling teams to proactively identify and resolve issues at specific sites, thus ensuring patient safety and data quality. Seamless Integration and Scalability A modern CDMS integrates with key systems such as Clinical Trial Management Systems (CTMS) and Randomization and Trial Supply Management (RTSM), establishing a unified data source, reducing data redundancy, and facilitating seamless trial scaling across various sites and regions. CUREXBIO: Your Partner in Precision Data Management Navigating modern clinical data complexities demands expertise beyond software, which is the essence of CUREXBIO. We recognize that high-quality data is crucial for successful clinical trials. Our comprehensive Clinical Data Management services leverage advanced CDMS technology to empower clients. Expertise System Selection & Implementation: We help you choose and implement the right CDMS platform for your specific trial needs. Tailored EDC Design: Our team builds intuitive and efficient electronic case report forms (eCRFs) that enhance site compliance and data accuracy. Careful Data Management: We don’t just manage data; we curate it. Our experts employ rigorous quality control processes to ensure your dataset is analysis-ready. Regulatory Readiness: We build compliance into every step, ensuring your data management processes will stand up to the strictest regulatory scrutiny. In the effort to expedite new therapies, effective data management acts as a crucial accelerator. Collaborating with a skilled data management team mitigates risks, optimizes resources, and enhances trial success prospects. For strategic advantages in clinical data, consider CUREXBIO’s advanced Clinical Data Management System for your next innovative breakthrough. Contact our data management team on bd@curexbio.com for effective and skilled data control.



