
End-to-End Clinical Research Services: From Preclinical Studies to Advanced Clinical Trial Management

In the dynamic realm of pharmaceutical and biotechnology innovation, the journey from drug or therapy concept to market involves intricate scientific validation, regulatory adherence, and coordinated efforts across several phases. End-to-end clinical research services, provided by Contract Research Organizations (CROs), play a crucial role in this process. These services minimize risks, speed up timelines, and ensure patient safety by offering comprehensive solutions, from early preclinical studies to clinical trial management and post-marketing surveillance. We at CurexBio, a clinical research services company, exemplify trusted partners in this sector, delivering integrated expertise to assist biotech, pharmaceutical, and medical device firms in transforming innovative ideas into effective treatments. End-to-End Clinical Research Services End-to-end clinical research services provide comprehensive support throughout the drug or device development lifecycle, unlike niche providers that focus on specific areas. Full-service CROs enable sponsors to either outsource the entire process or choose modular components for greater flexibility. Key phases typically include: This integrated approach promotes continuity, minimizes handoff errors, and adheres to global standards, including ICH-GCP, FDA, EMA, and CDSCO guidelines. The Journey: From Preclinical to Advanced Clinical Trials Preclinical Studies: Laying the Foundation Preclinical evaluation of a compound’s risks and benefits precedes human testing, involving in vitro assays, animal models for toxicology, safety pharmacology, and pharmacokinetics/pharmacodynamics (PK/PD) studies. A reliable Contract Research Organization (CRO) guarantees that these studies comply with Good Laboratory Practices (GLP) and yield substantial data for Investigational New Drug (IND) applications. Transition to Clinical Development Human trials commence after preclinical data is validated, facilitated by end-to-end services that include protocol development, regulatory strategy and submissions, and obtaining ethical approvals (IRB/IEC). Phase I-IV Clinical Trial Management Phases of clinical trial Advanced management encompasses site monitoring, patient recruitment strategies, real-time data oversight, and adaptive trial designs for efficient complexity handling. Additional Core Services CurexBio integral services Partnering with a dedicated CurexBio offers distinct advantages: CurexBio’s clinical research expertise CurexBio’s team of experts delivers precision, integrity, and collaboration, making them an ideal partner for companies seeking reliable end-to-end support. The evolving landscape of clinical research faced challenges such as increasing costs, demanding regulations, and the necessity for diverse patient populations. It emphasizes the need of experienced CROs like CurexBio, which enable sponsors to prioritize innovation while ensuring trials remain efficient, ethical, and successful. Explore CurexBio’s services on www.curexbio.com for a more effective transition from preclinical discovery to market success, promoting the core message of transforming scientific advancements into practical solutions through clinical trials.
What Is Clinical Trial Data Management and Why Is It Critical for Research Success?

Clinical trial data management (CDM) is essential in pharmaceutical and biotechnology research, enabling the effective handling of extensive data from clinical trials that evaluates the safety and efficacy of new treatments. CDM involves the systematic collection, cleaning, validation, and integration of data obtained from clinical trials, aimed at generating results that are reliable, high-quality, and statistically valid. The CDM process includes the design of data collection instruments, particularly electronic Case Report Forms (eCRFs), and emphasizes maintaining data integrity through strict quality control measures and adherence to regulatory standards. Key activities in Clinical Trials Data Management include: Data Collection: Gathering information from trial participants via standardized forms. Data Entry and Validation: Entering data into secure systems and running checks to identify discrepancies. Query Management: Resolving inconsistencies with trial sites. Database Lock: Finalizing the dataset for analysis once all queries are resolved. Modern Clinical Trial Data Management utilizes innovative tools such as Electronic Data Capture (EDC) systems. These advanced systems enhance efficiency by streamlining various processes and significantly minimizing errors when compared to conventional paper-based data collection methods. Need for Clinical Trial Data Management Critical for Research Success Poor data quality can significantly hinder the success of clinical trials, highlighting the critical need for effective Clinical Trial Data Management. Figure 1: Need for Clinical Trial Data Management Guarantee Data Integrity and Credibility : High-quality data is essential for accurate conclusions regarding treatment safety and efficacy, as errors or inconsistencies can undermine results, wasting resources and potentially endangering patients. Regulatory Compliance: Bodies like the FDA and EMA necessitate detailed data records for approvals, with robust CDM generating audit-ready datasets that adhere to GCP and CDISC guidelines. Increases Drug Development: Clean and validated data accelerates the process from trial completion to market approval by facilitating quicker statistical analysis and decision-making. Patient Safety: Timely identification of adverse events through proper data handling is essential for participant protection and informed risk-benefit assessments. Cost Efficiency: Preventing data-related delays and rework is crucial for saving expenses in multi-phase trials. Clinical Data Management transforms raw trial data into dependable evidence, promoting medical innovation. Partner with CurexBio for Expert Clinical Trial Data Management As a leading CRO, CurexBio provides comprehensive clinical trial data management solutions designed to expedite the drug development process, ensuring accuracy, compliance, and efficiency across all clinical trial phases. Advanced EDC systems with robust security and query management. End-to-end support from study design to database lock. Integration of cutting-edge technologies for real-time insights and regulatory adherence. CurexBio combines scientific expertise with operational excellence to assist biotech and pharmaceutical companies in managing complex trials. They provide audit-ready, high-quality data for both early-phase and large-scale global studies, facilitating successful outcomes. Ready to elevate your clinical research? Visit www.curexbio.com to learn more about CurexBio’s clinical trial data management and full-suite CRO services. Partner with us to turn your innovative therapies into reality, faster, safer, and with unwavering compliance.
Biostatistics Services and PK/PD Analysis: A Critical Foundation for Successful Clinical Trials
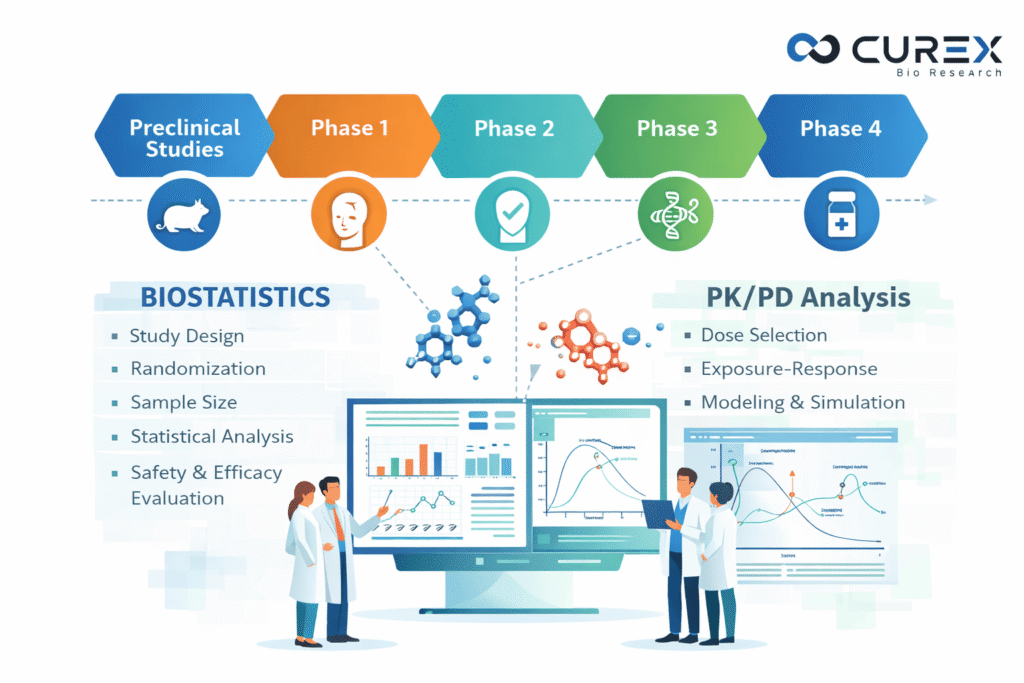
Biostatistics services play a pivotal role in drug development, where the transition from a promising molecule to an approved therapy requires rigorous evidence generation for safety and efficacy. Central to this process are biostatistics and pharmacokinetic/pharmacodynamic (PK/PD) analysis, which convert raw clinical data into actionable insights for informed decision-making in clinical trials. For researchers, pharmaceutical professionals, and stakeholders involved in the drug approval process, understanding these disciplines is essential. Additionally, CurexBio, a global Clinical Research Organization (CRO), provides comprehensive biostatistics services in clinical research to strengthen and accelerate drug development programs. The Phases of Drug Development and Clinical Trials Drug development involves a multi-stage process, where clinical trials are divided into phases to evaluate new therapies systematically. Figure 1: CurexBio’s Clinical Trial Process Biostatistics and pharmacokinetics/pharmacodynamics (PK/PD) are essential for making data-driven decisions throughout various phases. Biostatistics in Clinical Trials Biostatistics uses statistical methods to analyze biological and medical data, ensuring that clinical trial results are reliable, unbiased, and scientifically interpretable. It supports critical activities such as study design, sample size determination, randomization, data analysis, and interpretation of safety and efficacy outcomes. These statistical approaches are aligned with regulatory expectations outlined by the U.S. Food and Drug Administration, as detailed in the FDA guidance on statistical principles for clinical trials. Key roles include: Figure 2: Biostatistics Application in Clinical Trial Visualizations are essential in biostatistics, exemplifying how data is effectively presented in clinical trials. Without robust biostatistics, trials risk flawed conclusions, wasted resources, or regulatory rejection. PK/PD Analysis: Figure 3: CurexBio’s PK/PD modeling Role of PK/PD analysis in drug development PK/PD modeling associates drug exposure with its therapeutic effect, allowing for predictions of optimal dosing strategies. Figure 4: Classic PK/PD curves illustrate these relationships Biostatistics and PK/PD Essential in Drug Development These tools are essential for reducing risks, optimizing designs, and providing necessary evidence for regulatory approvals in pharmaceutical development. With robust pharmacokinetic/pharmacodynamic (PK/PD) data, predictions about human responses can be made from preclinical studies, while biostatistics ensures that statistical significance is meaningful in clinical contexts. The combined use of these methodologies effectively lowers attrition rates, accelerates timelines, and ultimately facilitates the faster delivery of safer therapies to patients. CurexBio Supports Your Clinical Trials CurexBio with facilities in India, the USA, and Canada, offers comprehensive clinical research services. Their expertise encompasses critical areas such as biostatistics and pharmacokinetics/pharmacodynamics (PK/PD) analysis. Our offerings include: Figure 5: CurexBio Biostatistics Services With extensive knowledge in various therapeutic areas, CurexBio provides biostatistics services that guarantee compliance, precision, and innovation, facilitating the process from protocol development to regulatory submission. We offers top-tier biostatistics and pharmacokinetics/pharmacodynamics (PK/PD) expertise, aimed at enhancing clinical trials.
Signal Detection in Drug Safety: The Modern Tools, Techniques, and Trends Shaping Patient Safety

In the rapidly evolving pharmaceutical sector, the safety profile of a drug continually develops post-regulatory approval, necessitating ongoing signal detection as a cornerstone of pharmacovigilance (PV). This process converts raw data on potential side effects into actionable intelligence that safeguards patients and supports clinical decision-making. The blog discusses contemporary methodologies enhancing signal detection, highlights the challenges faced by professionals, and outlines key trends such as artificial intelligence (AI) and real-world evidence (RWE) that are increasingly making drug safety surveillance proactive and predictive. What is a Safety Signal? At its core, a safety signal refers to information that indicates a new, potentially causal relationship between a medicine and an adverse event, or a novel aspect of an established association. It is important to emphasize that a safety signal is a hypothesis requiring further investigation, rather than a confirmed risk. Signals can manifest in various ways, such as an unknown adverse reaction, a known reaction occurring in a new population (e.g., the elderly), or an event reported with unusual frequency or intensity. The comprehensive process of collecting, monitoring, and evaluating these signals is termed signal management. Core Tools & Statistical Techniques Signal detection in pharmacovigilance involves the thorough examination of data from various sources, chiefly spontaneous reporting systems such as the FDA’s FAERS and the UK’s Yellow Card scheme, along with an increasing reliance on real-world data (RWD) obtained from electronic health records and claims databases. To effectively manage and analyze millions of data entries, safety scientists employ various statistical methods. A key technique in this process is disproportionality analysis, which serves to determine whether a particular drug-event combination occurs with greater frequency than would be statistically expected when compared to other drugs and events within the database. Technique Category Key Principle Proportional Reporting Ratio (PRR) Frequentist Compares the proportion of reports for a specific drug-event pair to the proportion for that event with all other drugs Reporting Odds Ratio (ROR) Frequentist Calculates the odds of an event being reported for a target drug versus all other drugs Information Component (IC) Bayesian Uses a Bayesian model to measure the disproportionality between observed and expected reporting rates Gamma Poisson Shrinker (GPS) Bayesian Employs Bayesian “shrinkage” to improve stability, especially for infrequent events No single method is universally superior; the most effective PV systems integrate multiple methodologies and combine statistical findings with detailed clinical review. The Future of Signal Detection & Emerging Trends The field is experiencing a major change influenced by advancements in technology and changes in regulations. AI & Machine Learning: AI enhances data processing and analysis, facilitating real-time monitoring and rapid pattern recognition. Machine learning models can predict safety issues earlier in drug development, and studies indicate that self-controlled designs combined with machine learning can lead to improved performance in specific situations. Real-World Evidence (RWE): Routinely collected healthcare data, such as EHRs, is increasingly being utilized alongside traditional spontaneous reports to enable quicker drug surveillance in wider and more diverse populations. Proactive & Predictive Surveillance: The shift is moving from a reactive model, which waits for reports, to a proactive system that anticipates risks by analyzing integrated data streams throughout a product’s lifecycle. Global Regulatory Modernization: Regulatory agencies such as the FDA and EMA advocate for updated methods that prioritize structured decision-making, enhanced documentation, and the application of advanced analytics. Despite the availability of advanced tools, key challenges persist in managing diverse, unstructured data from global sources. Ensuring data quality and integration is vital for timely decision-making, which is crucial for patient safety. Additionally, navigating global regulatory compliance demands ongoing vigilance and expertise. Addressing these challenges necessitates a strategic combination of technology, standardized processes, and extensive regulatory knowledge. CurexBio Supports Robust Signal Detection At CurexBio, we leverage specialized expertise and technology to transform industry trends into operational strengths through our signal detection services. Integrate and Analyze Diverse Data: We unify data from clinical trials, spontaneous reports, literature, and real-world data (RWD) to create a coherent safety picture. Implement Advanced Methodologies: We utilize appropriate tools, ranging from validated disproportionality analyses to predictive AI models, tailored to your specific needs. Ensure Regulatory Confidence: Our experts optimize signal management processes to align with FDA, EMA, and global regulatory standards, facilitating agile and compliant decision-making. Signal detection is crucial for patient safety, especially in the context of big data and advanced therapies. A proactive approach is essential to protect patients and support the success of pharmaceutical innovations. CurexBio (contact us on bd@curexbio.com) offers tailored pharmacovigilance solutions to enhance drug safety surveillance.
Using Real-World Evidence (RWE) to Support Smarter Clinical and Regulatory Decisions

For decades, the randomized controlled trial (RCT) has served as the primary standard for establishing a treatment’s efficacy. Nonetheless, an important question persists post-approval regarding the drug’s performance among the diverse patient population encountered in routine clinical settings. Real-World Evidence (RWE) has emerged as a crucial tool, offering significant insights that are reshaping the evaluation processes employed by regulators, payers, and pharmaceutical sponsors throughout the treatment’s lifecycle. What Exactly is Real-World Evidence? Real-World Evidence (RWE) is fundamentally clinical evidence obtained through the examination of Real-World Data (RWD), which encompasses data gathered routinely from diverse healthcare sources beyond the rigid parameters of clinical trials. Common sources of RWD include: Electronic Health Records (EHRs): Data from doctor’s visits, hospital stays, and diagnoses. Claims and Billing Data: Information on prescriptions, procedures, and insurance coverage. Disease or Product Registries: Longitudinal databases tracking patients with specific conditions or treatments. Patient-Generated Data: Information from wearables, home monitoring devices, and patient-reported outcomes. When analyzed to address specific clinical questions, such as the long-term safety or effectiveness of a treatment within a wider population, raw RWD is transformed into comprehensive RWE. Bridging the Gap: How RWE Complements Traditional Trials RCTs and RWE complement each other rather than compete. RCTs excel in demonstrating efficacy within controlled environments, while RWE provides insights into effectiveness within the complexities of everyday clinical practice. The table below highlights their complementary strengths: Feature Randomized Controlled Trial Real-World Evidence Primary Purpose Assess Efficacy Demonstrates effectiveness Setting Highly controlled, experimental Routine world clinical practice Patient Population Strict eligibility criteria; often homogeneous Diverse, heterogeneous Treatment Pattern Fixed, as per protocol Variable, at physician’s and patient’s discretion Key Strength High internal validity; controls for bias via randomization High external validity; shows real-world applicability and long-term outcomes This partnership addresses the “efficacy-effectiveness gap” by offering a comprehensive assessment of a product’s value throughout its lifecycle, from pre-approval stages to post-market surveillance. How Regulators Use RWE: From Safety Monitoring to New Approvals Regulatory agencies such as the U.S. Food and Drug Administration (FDA) and the European Medicines Agency (EMA) have historically utilized RWE for post-market safety monitoring. Notable initiatives like the FDA’s Sentinel System leverage extensive healthcare databases to actively identify and evaluate safety signals associated with approved medications. Currently, the function of RWE is swiftly evolving to encompass more active roles in regulatory decision-making. This shift is largely influenced by initiatives like the 21st Century Cures Act, which encourages regulators to accept Real-World Evidence (RWE) as a basis for supporting new indications for existing drugs or fulfilling requirements for post-approval studies. In Europe, a study examining EMA approvals from 2020 to 2023 revealed that Real-World Evidence (RWE) is increasingly relevant, especially in oncology, where it constitutes 37.5% of RWE studies. However, it has only been integrated into fewer than 10% of new marketing authorizations, suggesting there is considerable potential for expansion in this area. For pharmaceutical and biotech companies, RWE serves as a strategic asset, guiding decisions throughout the entire lifecycle of a product. Clinical Development: Real-World Evidence (RWE) can optimize trial design, identify potential study sites, and help create external control arms for diseases where randomized trials are unethical or impractical. Regulatory Submissions: As shown above, RWE can provide confirmatory or pivotal evidence to support new drug applications or label expansions, especially in rare diseases. Market Access & Commercialization: Payers demand evidence of real-world effectiveness and cost-effectiveness, with Real-World Evidence (RWE) showcasing a product’s value across diverse populations, thereby aiding reimbursement and market access strategies. Pharmacovigilance & Safety: RWE supports large-scale safety monitoring of products, enhancing risk management and ensuring patient safety. Navigating the challenges of generating regulatory-grade RWE involves ensuring data quality, consistency, and fitness for purpose, along with addressing issues of data privacy and potential biases. Success relies on a rigorous, transparent approach that includes selecting appropriate data sources and study designs, employing robust statistical methods, and following best practices and regulatory guidance. The future of evidence is moving towards an integrated strategy, merging the controlled environment of RCTs with the insights gained from RWE, necessitating expertise in data science, epidemiology, regulatory science, and therapeutic areas. CurexBio specializes in converting complex real-world data into regulatory-grade evidence, offering end-to-end RWE services. Our services assist with study design, data sourcing, advanced analytics, and regulatory submission support to showcase the true value of treatments. Contact CurexBio to explore how our RWE solutions can enhance clinical and commercial goals.
Avoid These 5 Costly CSR and Submission Errors with Expert Medical Writing Services

Experts consistently identify recurring issues that trigger regulatory delays and queries. Engaging experienced medical writing services helps reduce these risks by ensuring CSRs and submissions are accurate, well-structured, and aligned with regulatory expectations. Mistake /Error Category Common Examples & Consequences How to Avoid Poor Narrative & Lack of Clarity Failing to weave data into a logical scientific story; overusing jargon. Write clear, regulator-friendly text that interprets data for patient impact Data Inconsistencies & Integrity Issues Numbers mismatching across documents (CSR, SAP, etc.); poor data integrity can lead to FDA rejecting entire datasets Implement rigorous cross-document QC and validation. Use validated systems and risk-based monitoring. Ignoring Guidelines & Formatting Errors Deviating from ICH E3 (CSR structure) or eCTD formatting rules Use updated checklists and expert writers versed in current standards Weak Data Collection & Management Using non-validated tools (spreadsheets) or paper systems; creates compliance risks and errors. Use pre-validated Electronic Data Capture (EDC) and integrated data systems. Reactive Process & Poor Collaboration Writing the CSR only after trial ends; siloed teams cause delays and disorganization Involve regulatory expertise early; build structured cross-functional review processes How Curex Bio’s Services Help You Avoid These Mistakes CurexBio offers specialized services — including expert medical writing services — that directly target the root causes of common submission errors. To Ensure Data Quality & Integrity (Mistakes 2 & 4): Our clinical monitoring utilizes Source Data Verification (SDV) and risk-based monitoring strategies, which involve a combination of on-site visits and remote oversight to proactively detect and address data discrepancies. Additionally, the monitoring framework includes central laboratory support with integrated data systems aimed at ensuring a standardized, accurate, and traceable flow of data — forming a strong foundation for high-quality medical writing services and regulatory submissions. To Build a Compliant Submission Foundation (Mistakes 1, 3 & 5): CurexBio oversees comprehensive clinical operations, which encompass site selection, training, and careful maintenance of regulatory files. This approach guarantees that trials adhere to Good Clinical Practice (GCP) standards from initiation, establishing a trustworthy basis for the final report. Our services include comprehensive pharmacovigilance and safety monitoring, essential for the safety sections of any clinical submission, thereby supporting the broader clinical narrative. Key Considerations for a Successful Submission Beyond engaging expert partners, keep these principles in mind: Begin with Preplanned: Effective regulatory strategies start with proactive planning during protocol development, which avoids hasty and error-prone writing later on. Data integrity importance: Data integrity is essential and should be considered non-negotiable, as emphasized by the FDA. Significant issues can result in total data rejection, leading to substantial time and resource losses. Test Your Processes: Before the trial begins, simulate data collection and workflow with site staff to identify practical hurdles that may affect data quality later. For detailed insights into specific therapeutic areas or phases of development, it is recommended to explore CurexBio’s Clinical Development and Clinical Monitoring services. If you are in the process of planning a trial or preparing a submission, our expert medical writing services can support you in addressing documentation and data management challenges to ensure high-quality, compliant submissions.



